


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
城戸 健太朗; 金子 政志
Journal of Computational Chemistry, 44(4), p.546 - 558, 2023/02
被引用回数:1 パーセンタイル:13.58(Chemistry, Multidisciplinary)Distribution of solvent molecules near transition-metal complex is key information to comprehend the functionality, reactivity and so on. However, polarizable continuum solvent models still are the standard and conventional partner of molecular-orbital (MO) calculations in the solution system including transition-metal complex. In this study, we investigate the conformation, hydration structure and ligand substitution reaction between NO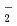 and H
and H O in aqueous solution for [Ru(NO)(OH)(NO)
O in aqueous solution for [Ru(NO)(OH)(NO)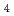 ]
]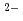 (
(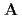 ), [Ru(NO)(OH)(NO)
), [Ru(NO)(OH)(NO)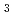 (ONO)] (
(ONO)] (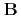 ) and [Ru(NO)(OH)(NO)(HO)]
) and [Ru(NO)(OH)(NO)(HO)]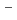 (
( ) using a combination method of MO theories and a state-of-the-art molecular solvation technique (NI-MC-MOZ-SCF). In the complexes, the treatment is essentially required because except for nitrosyl ligand, a strong hydrogen bond is formed between the ligand and solvent water. These results are complementary to the data previously obtained by
) using a combination method of MO theories and a state-of-the-art molecular solvation technique (NI-MC-MOZ-SCF). In the complexes, the treatment is essentially required because except for nitrosyl ligand, a strong hydrogen bond is formed between the ligand and solvent water. These results are complementary to the data previously obtained by 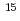 N NMR experiment. A dominant species is found in the complex conformers and, as expected, different between the solvent models, which reveals that molecular solvation beyond continuum media treatment are required for a reliable description of solvation near transition-metal complex. In the stability constant evaluation of ligand substitution reaction, similar to the previous reports, an assumption that considers the direct association between the dissociated nitrite anion and complex is useful to obtain a reliable stability constant.
N NMR experiment. A dominant species is found in the complex conformers and, as expected, different between the solvent models, which reveals that molecular solvation beyond continuum media treatment are required for a reliable description of solvation near transition-metal complex. In the stability constant evaluation of ligand substitution reaction, similar to the previous reports, an assumption that considers the direct association between the dissociated nitrite anion and complex is useful to obtain a reliable stability constant.
志賀 基之
Journal of Computational Chemistry, 43(27), p.1864 - 1879, 2022/10
被引用回数:3 パーセンタイル:40.16(Chemistry, Multidisciplinary)経路積分法に基づいて、量子振動動力学のための新たな近似法(Brownian chain molecular dynamics: BCMD)を提案する。セントロイド分子動力学法やリングポリマー動力学法などの従来法では、量子振動スペクトルが低温条件で正しく計算できない。そこで、本研究では、その問題の源である非物理的な共鳴や振動回転カップリングを抑制するため、原子の非重心自由度に対して過減衰ランジュバン方程式を導入する。軽水や重水などの赤外吸収スペクトルへの応用例を通じて、この近似を検証する。また、BCMD法を電子状態理論と組み合わせた第一原理シミュレーションを実証する。
Pal, A.*; Pal, S.*; Verma, S.*; 志賀 基之; Nair, N. N.*
Journal of Computational Chemistry, 42(28), p.1996 - 2003, 2021/10
被引用回数:8 パーセンタイル:54.02(Chemistry, Multidisciplinary)温度加速断片サンプリングは、高次元の自由エネルギー地形の計算法である。従来法では、重み付きヒストグラム解析法を用いて計算の後処理を行うが必要があった。この研究では、平均力を使用して後処理を行わずに済む洗練された手法を確立する。これを用いると、アラニンジペプチドとトリペプチドの2次元および4次元の自由エネルギー地形が約kcal/molの誤差内で計算されること実証した。この方法は、計算化学における自由エネルギー計算において幅広い応用が期待される。
近藤 友美; 佐々木 岳彦*; 志賀 基之
Journal of Computational Chemistry, 42(25), p.1783 - 1791, 2021/09
被引用回数:1 パーセンタイル:6.39(Chemistry, Multidisciplinary)高温水溶液中の糖アルコール脱水反応は、有機溶媒を使用せずに環境に優しいバイオマス変換を可能にする重要な反応である。本研究では、高温酸性水中でのソルビトール(SBT)の脱水反応のメカニズムを理解するため、第一原理および半経験的電子状態理論に基づくメタダイナミクス(MTD)シミュレーションによる自由エネルギー解析を行った。反応機構は2分子求核置換型(S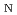 2)であり、それによって中間体として生成されたヒドロキシル基のプロトン化の自由エネルギーが酸性種の影響を受けることがわかった。また、5員エーテル生成物につながる反応経路の自由エネルギー障壁は、6員エーテル生成物につながるものよりも低いことがわかった。
2)であり、それによって中間体として生成されたヒドロキシル基のプロトン化の自由エネルギーが酸性種の影響を受けることがわかった。また、5員エーテル生成物につながる反応経路の自由エネルギー障壁は、6員エーテル生成物につながるものよりも低いことがわかった。
近藤 友美; 佐々木 岳彦*; Ruiz-Barragan, S.*; Ribas-Ari o, J.*; 志賀 基之; Ruiz-Barragan, S.*
o, J.*; 志賀 基之; Ruiz-Barragan, S.*
Journal of Computational Chemistry, 42(3), p.156 - 165, 2021/01
被引用回数:4 パーセンタイル:20.61(Chemistry, Multidisciplinary)本研究では、固定されたバイアスポテンシャル下の正準サンプリングによって、メタダイナミクスを改良する新たな計算手法を提案した。この手法は、2つ以上の自由エネルギー障壁を異なる条件または競合する条件での化学反応間で比較する場合などに役立つ。この方法を用いて、高温水溶液中の多価アルコール脱水反応の酸依存性を調べた。その結果、酸の種類によらず、この反応はS2メカニズムを介して進行することがわかった。一方、中間体として生成されるヒドロキシル基のプロトン化における自由エネルギー変化が、酸の種類によって影響を大きく受けることがわかった。
城戸 健太朗
Journal of Computational Chemistry, 40(24), p.2072 - 2085, 2019/09
被引用回数:3 パーセンタイル:13.02(Chemistry, Multidisciplinary)Mean-field treatment of solvent provides an efficient technique to investigate chemical processes in solution in QM/MM framework. In the algorithm, an iterative calculation is required to obtain the self-consistency between QM and MM regions, which is a time-consuming step. In the present study, we have proposed a non-iterative approach by introducing a linear response approximation (LRA) into the solvation term in the one-electron part of Fock matrix in a hybrid approach between MO calculations and a three-dimensional integral equation theory for molecular liquids (MC-MOZ-SCF; Kido 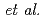 , J. Chem. Phys.
, J. Chem. Phys. 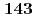 , 014103 (2015)). To save the computational time, we have also developed a fast method to generate electrostatic potential map near solute and the solvation term in Fock matrix, using Fourier transformation (FT) and real spherical harmonics expansion (RSHE). To numerically validate the LRA and FT-RSHE method, we applied the present approach to water, carbonic acid and their ionic species in aqueous solution. Molecular properties of the solutes were evaluated by the present approach with four different types of initial wave function and compared with those by the original (MC-MOZ-SCF). From the averaged speed up ratio, the present approach is 13.5 times faster than MC-MOZ-SCF.
, 014103 (2015)). To save the computational time, we have also developed a fast method to generate electrostatic potential map near solute and the solvation term in Fock matrix, using Fourier transformation (FT) and real spherical harmonics expansion (RSHE). To numerically validate the LRA and FT-RSHE method, we applied the present approach to water, carbonic acid and their ionic species in aqueous solution. Molecular properties of the solutes were evaluated by the present approach with four different types of initial wave function and compared with those by the original (MC-MOZ-SCF). From the averaged speed up ratio, the present approach is 13.5 times faster than MC-MOZ-SCF.
櫻庭 俊; 松林 伸幸*
Journal of Computational Chemistry, 35(21), p.1592 - 1608, 2014/08
被引用回数:55 パーセンタイル:79.95(Chemistry, Multidisciplinary)ERmodはエネルギー表示法に基づく、効率よく溶媒和自由エネルギーを近似計算するソフトウェアである。溶液系・参照溶媒系の2状態での分子シミュレーションのみから、溶媒和自由エネルギーを計算することが可能である。ERmodのサブプログラムである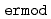 は溶質溶媒間相互作用エネルギーの分布関数を計算し、別のサブプログラムである
は溶質溶媒間相互作用エネルギーの分布関数を計算し、別のサブプログラムである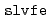 は溶媒和自由エネルギーを分布関数から溶液の近似汎関数理論に従い計算する。本論文ではERmodの設計と実装について記述し、またそのパフォーマンスを2つの有機分子と2つのタンパク質を溶質とする系について示す。ERmodの利用範囲は幅広く、均一溶媒系である流体やポリマーのみならず、ミセルや脂質膜系などのナノ不均一系まで適用可能である。ERmodは
は溶媒和自由エネルギーを分布関数から溶液の近似汎関数理論に従い計算する。本論文ではERmodの設計と実装について記述し、またそのパフォーマンスを2つの有機分子と2つのタンパク質を溶質とする系について示す。ERmodの利用範囲は幅広く、均一溶媒系である流体やポリマーのみならず、ミセルや脂質膜系などのナノ不均一系まで適用可能である。ERmodは にて公開されている。
にて公開されている。
池部 仁善; 櫻庭 俊; 河野 秀俊
Journal of Computational Chemistry, 35(1), p.39 - 50, 2014/01
被引用回数:14 パーセンタイル:42.16(Chemistry, Multidisciplinary)A novel, efficient sampling method for biomolecules is proposed. The partial McMD was recently developed as a method that improved generalized ensemble (GE) methods to focus sampling only on a part of a system (GEPS); however, it was not tested well. We found that partial McMD did not work well for poly-lysine decapeptide and gave significantly worse sampling efficiency than a conventional GE. Herein, we elucidate the fundamental reason for this and propose a novel GEPS, adaptive lambda square dynamics (ALSD), which can resolve the problem faced when using partial McMD. We demonstrate that ALSD greatly increases the sampling efficiency over a conventional GE. We believe that ALSD is an effective method and is applicable to the conformational sampling of larger and more complicated biomolecule systems.
藤本 浩文*; 樋口 真理子; 小池 学*; 大出 裕高*; Pinak, M.; Kotulic Bunta, J.*; 根本 俊行*; 作道 隆*; 本田 尚子*; 前川 秀彰*; et al.
Journal of Computational Chemistry, 33(3), p.239 - 246, 2012/01
被引用回数:36 パーセンタイル:68.39(Chemistry, Multidisciplinary)リジン残基のアセチル化は、タンパク質が受ける翻訳後修飾の中で最も一般的なものの一つである。本研究では、DNA修復酵素の一つであるKuタンパク質内のリジン残基がアセチル化されると、その基質であるDNAとの結合力がどのように変化するのかをコンピュータシミュレーションを用いて検証した。擬似アセチル化タンパク質のモデルとして実験的によく用いられる、グルタミン置換変異体(KQ変異体)ではDNAとの結合力が下がり、非擬似アセチル化タンパク質モデルとして用いられるアルギニン置換変異体(KR変異体)では結合力は低下しなかった。このシミュレーション結果は既報の実験結果と完全に一致している。一方、これらのリジン残基をアセチル化してもDNAとの結合力は低下しなかった。したがって、擬似アセチル化タンパク質のモデルとしてKQ変異体を用いると、アセチル化の影響を過大評価する場合があると考えられる。
石田 恒
Journal of Computational Chemistry, 31(12), p.2317 - 2329, 2010/09
Branch migration of the Holliday junction takes place at the center of the RuvA tetramer. To elucidate how branch migration occurs, umbrella sampling simulations were performed for complexes of the RuvA tetramer and Holliday junction DNA. While conventional umbrella sampling simulations set sampling points a priori, the umbrella sampling simulation in this study set the sampling points one by one in order to search for a realistic path of the branch migration during the simulations. Starting from the X-ray structure of the complex, in which the hydrogen bonds between two base-pairs were unformed, the hydrogen bonds between the next base-pairs of the shrinking stems were observed to start to disconnect. At the intermediate stage, three or four of the eight unpaired bases interacted closely with the acidic pins from RuvA. During the final stage, these bases moved away from the pins and formed the hydrogen bonds of the new base-pairs of the growing stems. The free-energy profile along this reaction path showed that the intermediate stage was ameta-stable state between two free-energy barriers of about 10-15 kcal/mol. These results imply that the pins play an important role in stabilizing the interactions between the pins and the unpaired base-pairs.
藤本 浩文*; Pinak, M.; 根本 俊行*; O'Neill, P.*; 久米 悦雄; 斎藤 公明; 前川 秀明*
Journal of Computational Chemistry, 26(8), p.788 - 798, 2005/06
被引用回数:23 パーセンタイル:57.5(Chemistry, Multidisciplinary)電離放射線によるDNAクラスター損傷は修復され難く、細胞のガン化など生体にとって深刻な事態を引き起こす原因の一つと考えられている。DNAクラスター損傷を持つDNA分子には、単独の損傷を持つ分子と比べるとDNA修復酵素が作用し難いことが、生化学的・分子生物学的実験によって示されているが、どのような要因が酵素の作用阻害に関わっているかはいまだ不明である。そこで本研究では、DNAクラスター損傷における酵素の作用阻害の要因を、計算科学的手法を用いて考察した。既報の実験で用いられたDNA分子と同配列となるように、7,8-dihydro-8-oxoguanine(8-oxoG)及びapurinic/apyrimidinic(AP) siteという2つの酸化損傷部位が数塩基はなれた位置に存在する40merのDNA分子を、2損傷部位間の距離を変えて6種類設計し、それぞれに対し分子動力学的(MD)シミュレーションを1nsのオーダーで行った。その結果、損傷部位における分子の屈曲や、損傷塩基と相補鎖上の塩基との相互静電エネルギーの減少など損傷DNA分子に特徴的な構造や性質が観察された。これらの特徴によって修復酵素がDNAに結合できず、したがって修復効率が低下したのではないかと推察される。
Pinak, M.
Journal of Computational Chemistry, 24(7), p.898 - 907, 2003/04
被引用回数:8 パーセンタイル:36.11(Chemistry, Multidisciplinary)突然変異を誘発するDNA酸化損傷である7,8-ジヒドロ-8-オキソグアニン(8-oxoG)と、ヒト修復酵素であるオキソグアニングリコシレース1(hOGG1)とが形成する複合体の分子動力学的シミュレーションを1ナノ秒(ns)行い、DNA-酵素複合体の形成にかかわる動力学的過程の検討を行った。分子動力学的シミュレーション開始後500ピコ秒(ps)でDNA-酵素複合体が形成され、シミュレーションが終了する1ns後まで安定していた。複合体はおもにDNAと酵素のファンデルワールス面の重なり合いによって定義されており、アルギニン313のN末端はヌクレオチドのリン酸ジエステル結合の近傍に位置して、8-oxoGはアミノ酸と損傷と化学反応を可能にしている。また水素結合の切断によって部分的に構造が破壊されたB型DNAが観察された。8-oxoGの5'位のリン酸ジエステル結合は、アルギニン313のN末端に近い位置に移動している。DNAと酵素の近接箇所では水分子を介した水素結合が形成され、複合体の安定性を高めている。正常なDNAを用いた同様の分子系で行ったシミュレーションでは、複合体や水分子を介した水素結合は観察されたなかった。
Pinak, M.
Journal of Computational Chemistry, 22(15), p.1723 - 1731, 2001/11
被引用回数:3 パーセンタイル:21.86(Chemistry, Multidisciplinary)チミングリコール(TG)を持つDNAと修復酵素エンドヌクレアーゼIIIの複合体形成過程について、分子動力学計算を用いて調べた。修復酵素とTGを持つ30塩基対長のDNAが水溶液中に存在する系をモデル化し、2ナノ秒間のシミュレーションを行った。シミュレーション開始から約1ナノ秒後にDNAと修復酵素は複合体を形成し、シミュレーションが終了するまで安定な構造を保持した。酵素とDNAの結合領域において、グルタミン酸がリン酸結合のC3'分子から1.6オングストロームの位置まで接近していることがわかった。これは、修復過程で切断される2つの結合のうちの1つにあたる。また、TGのある部分でDNAは折れ曲がったが、この変形により修復酵素が損傷部分に近づきやすくなると考えられる。さらに、静電エネルギーの変化も損傷認識過程において重要な寄与をしていることが確認された。

